SparkMap: Scalable parallel genome mapping in a distributed computing architecture
Abstract
Background: Many high-throughput sequencing (HTS) techniques require mapping reads to a reference genome. To accommodate the rapid growth in the size of HTS libraries, innovative mapping algorithms have been employed that greatly reduce genome alignment times. Nevertheless, mapping reads to reference genomes is computationally intensive and often represents a rate-limiting step for analysis of HTS libraries.
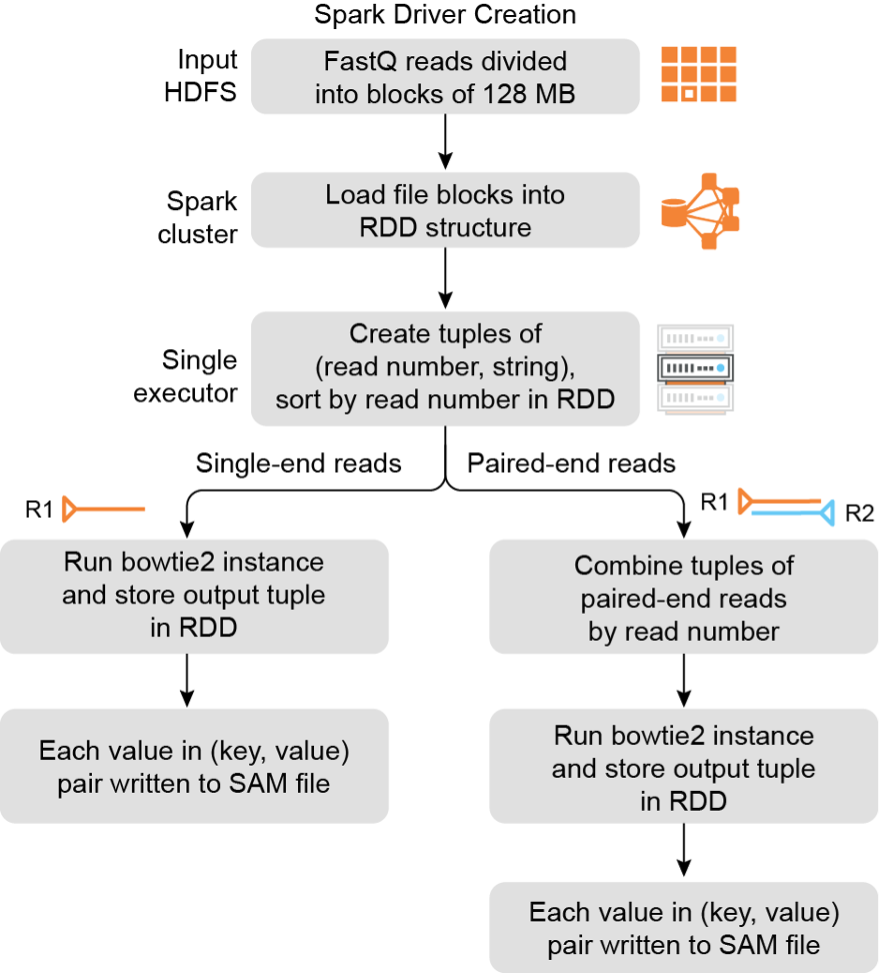
Results: Here we provide an efficient mapping framework, SparkMap. SparkMap is implemented by coupling existing mapping software to the Apache Spark distributed data processing framework and maintains identical accuracy as the underlying mapping software. We compared the performance of SparkMap and Bowtie2 based on variable numbers of cores and RAM, and report how scaling these resources influence performance. Furthermore, we compared the mapping speeds of conventional Bowtie2, HISAT2, and BBMAP against SparkMap configured to use these mappers, to show the generalizability of the framework. To benchmark upper limits of performance gains, we compared a reference 2-core configuration using Bowtie2, HISAT2, or BBMAP, to a 165-core SparkMap configuration based on the same mappers.
Conclusions: We found the 165-core SparkMap configuration achieves mapping times 2, 16, and 33-fold faster than the respective 2-core reference configuration. By enabling the utilization of large numbers of CPU cores for mapping, SparkMap is able to process large volumes of HTS data in parallel using a distributed computing cluster. SparkMap is scalable with regards to both the size of a dataset and the number of available compute nodes.
Presented at the International Society for Computational Biology ROCKY 2019 Conference